Introdução
As doenças do sangue também chamadas de doenças hematológicas ou Hemopatias acometem os sistemas eritrocitarios, leucocitários, plaquetarios e da coagulação.
Estas doenças provocam alterações que modificam o funcionamento normal do organismo, afetando o carregamento de oxigênio e os nutrientes necessários ao funcionamento celular; a eliminação das excretas metabólicas e o mecanismo de coagulação do sangue.
Na pratica de fisioterapia, os distúrbios sanguíneos apresentam sintomas comumente relacionados ao uso de drogas antiinflamatórias, complicações da quimioterapia ou da radiação e complicações neurológicas associadas com anemia perniciosa.O fisioterapeuta deve ficar alerta sobre a possibilidade de um distúrbio hematológico sistêmico, em casos de alterações nas mãos ou nos leitos das unhas; sintomas de dispnéia, fraqueza, fadiga e palpitações; sintomas neurológicos, tais como dor de cabeça, sonolência, tontura, síncope, polineuropatia; e fácil contusão ou sangramento.(GOODMAN & SNDYNER, 2002).
Para conhecer as hemopatias e as alterações causadas por elas, é necessário conhecer o tecido sanguíneo, sua importância e funções principais.
1. O Sangue
O sangue é o tecido conjuntivo líquido que circula pelo sistema vascular através do coração, das artérias, dos capilares e das veias.Produzido na medula ósseas dos ossos chatos, vértebras, quadril, crânio e esterno através da hematopoese, o sangue é constituído por dois componentes principais: o Plasma e os elementos corpusculares ou Figurados.
1.1 O Plama é a parte líquida do sangue de coloração amarelo-claro ou cinza-amarelado, composto por água (90%), proteínas e sais minerais, através do qual circulam substâncias nutritivas como proteínas, enzimas, hormônios, imunoglobulinas, albumina e fatores de coagulação, necessárias a vida das células.O plasma representa 55% do volume do sangue circulante.
1.2 Os Elementos Figurados ou Corpusculares são distribuídos em:
Eritrócitos (glóbulos vermelhos): Células em forma de disco que possuem Hemoglobina (pigmento no qual se fixa o oxigênio) e que transportam o oxigênio aos tecidos e movem o dióxido de carbono através deles.Os glóbulos vermelhos são produzidos pela medula óssea, sobretudo no esterno e nas costelas;
• Leucócitos (glóbulos brancos): São maiores em tamanho se comparados aos eritrócitos.Atuam nas respostas inflamatórias e imunes e são distribuídos em vários gêneros: os Eutrófilos, que atuam defendo o organismo das bactérias; os Linfócitos, atuantes nas defesas imunitárias; os Eosinófilos, que identificam os alergêneos e outros corpos estranhos; os Basófilos, que através de suas substâncias podem agir como coagulantes e atuar contra a inflamação dos vasos sanguíneos; e os Monócitos, que absorvem as bactérias. Os glóbulos brancos são produzidos na medula óssea e nos gânglios linfáticos, no baço, no timo e nas amígdalas;
• Plaquetas: possuem forma achata e são as menores das células sanguíneas.Junto com os fatores de coagulação do plasma, controlam a coagulação do sangue.
As substâncias nutritivas e oxigênio são distribuídos às células do organismo através da circulação. Além de transportar o oxigênio e atuar na remoção do dióxido de carbono, o sangue também possui papel importante nas defesas imunitárias do organismo.
2. Doenças do Sangue
As hemopatias podem ser adquiridas ou hereditárias, surgir nos próprios componentes do sangue ou nos processos que participam na formação das células sanguíneas.Podem ainda ser dividas em Primarias, secundarias e associadas.
2.1.Hemopatias Primarias: refere-se aos distúrbios relacionados aos sistemas eritrocitarios, leucocitários, plaquetarios e da coagulação.
2.2.Hemopatias Secundárias: é o termo utilizado quando a lesão principal não se localiza nos órgãos hemopoiéticos ou nas células sanguíneas, como por exemplo, na esquistossomose mansônica, onde por lesão hepática pode ocorrer alteração da volemia e dos fatores de coagulação.
2.3.Hemopatias Associadas pertencem ao quadro clinico do grupo de moléstias cujas causas podem ou não ser bem definidas.Isto ocorre, por exemplo, nas reticuloendotelioses de acumulo, que ocorre em algumas doenças hereditárias nas quais há deficiência de enzimas que atuam no metabolismo dos polissacarídeos e dos lipídeos.
3. Principais Doenças Do Sangue
3.1.Doenças Eritrocitárias
Os distúrbios envolvendo os eritrócitos podem ser classificados como:
• Anemia (Baixo número de eritrócitos);
• Policitemia (Aumento no número de eritrócitos);
• Pecilocitose (Eritrócitos com formas anormais);
• Anisocitose (variações anormais no tamanho dos eritrócitos);
• Hipocromia (Deficiência de hemoglobina).
3.1.1.Considerações sobre anemia
Anemias são as doenças do sangue mais freqüente.Refere-se a uma redução da capacidade do sangue de transportar oxigênio como resultado de uma anormalidade na quantidade ou qualidade dos eritrócitos.Não é uma doença, mas um sintoma de uma série de distúrbios sanguíneos diferentes. (GOODMAN & SNDYNER, 2002).
A anemia pode ser causada por um sangramento excessivo, pela diminuição da produção de eritrócitos ou pelo aumento de sua destruição (hemólise) e são classificadas de acordo com dois aspectos: Morfógico ou fisiopatológico.
3.1.2. Classificação morfológica das anemias:
Este critério dá a idéia do aspecto morfológico do eritrócito, e não da causa da anemia. Nesses grupos encontram-se:
• Macrocíticas e normocrômicas:presença de hemácias de grande volume e geralmente hipercrômicas.
• normocíticas e normocrômicas:são normocrômicas e incluen-se neste grupo as anemias hemoliticase as anemias aplasicas.
• microcíticas e hipocrômicas: predomínio de hemácias de pequeno volume e pobres em hemoglobina ou hipocrômicas.Neste grupo estão incluídas as anemis ferroprivas.
3.1.3.Classificação fisiopatológica das anemias:
Este critério fornece a base fisiopatológica para explicar os diferentes tipos de anemia.Podem ser assim classificadas por:
• Falta de produção de eritrócitos: podem ser decorrentes da deficiência de elementos essenciais á produção de glóbulos vermelhos do sangue como o ferro, ácido fólico, vitamina B12, proteínas, entre outras vitaminas; por deficiência dos eritroblastos; por infiltração medular; por endocrinopatias; por insuficiência renal crônica, dentre outras.
• Excesso de Destruição de eritrócitos: são as anemias que podem ser causadas por defeitos intrínsecos e extrínsecos dos eritrócitos;
• Perdas de sangue: causadas pelas perdas agudas ou crônicas do sangue.
3.1.4.Tipos de Anemia
As anemias mais freqüentes e de relevância tanto médica quanto social são:
• Anemia por Carência de ferro: anemia ferroprivas;
• Anemia por carência de vitamina B12: anemia perniciosa;
• Anemia das doenças crônicas;
• Anemias decorrentes de doenças de medula óssea aplastica, leucemias e tumores medulares;
• Anemias por agressão periférica aos eritrócitos: malaria, anemias hemolíticas imunológicas, anemia por fragmentação dos eritrócitos;
• Anemias por defeitos genéticos: talessemias, falciforme, esferocitose.
3.1.5 Sinais e Sintomas Clínicos das Anemias
Os sinais e sintomas mais comuns nas anemias são:
• Pressão sanguínea sistólica reduzida;
• Fadiga e fraqueza muscular;
• Dispnéia por esforço acompanhada por palpitações cardíacas e rápida pulsação (quando a anemia é mais severa);
• Pele com palidez (palma e leito das unhas) ou pele de coloração amarelada (mucosa conjuntiva).
3.1.6 Tratamento das Anemias
o tratamento das anemias é causal.A anemia por perda sanguínea, por exemplo, requer a investigação da hemorragia e em alguns casos, a intervenção cirúrgica acompanhada, em alguns casos, por transfusão de sangue.
3.1.7.Considerações sobre a Anemia Falciforme
A anemia falciforme é um termo utilizado para denominar um grupo de distúrbios autossômicos hereditários recessivos, que se caracterizam pela má formação das hemácias, que assumem forma semelhante a uma foice.
Esta mudança na forma da célula é resultado de uma substituição de aminoácido em hemoglobina, levando a esta se agregar em longas cadeias, alterando então o aspecto morfológico da célula.Com isso, a célula perde sua capacidade de deformação e acaba por forçar a passagem pelos pequenos vasos sanguíneos, e deste modo o tecido fica privado de um suprimento sanguíneo adequado.
3.1.8 Sinais e Sintomas da Anemia Falciforme:
o Dor de leve a intensa: torácica, articulações, abdome, dores de cabeça;
o Pés e mãos edemaciados;
o Icterícia (pele e mucosas amareladas);
o Infecções de repetição, particularmente pneumonias e meningites;
o Cálculos na vesícula (em idade jovem):
o Neurológicas:convulsões,sonolência,coma,parstesia,nistagmo,paralisias de nervos cranianos,perda visual,rigidez cervical;
o Renais: enurese, nocturia, hematuria, pielonefrite, necrose papilar renal;
o Dentre outras complicações.
3.1.9 Tratamento e Cuidados:
Ainda não existem medicamentos eficazes para o tratamento da anemia falciforme.O tratamento é voltado para a prevenção de complicações, onde os episódios dolorosos são tratados com a administração de analgésicos, líquidos e oxigênio.Dietas com suplementos de acido fólico,entre outras medidas são utilizadas.
4.COAGULAÇÃO DO SANGUE
O organismo conta com um mecanismo vital contra as perdas excessivas de sangue - a coagulação -, que auxilia na interrupção das hemorragias fechando os vasos sangüíneos abertos e, portanto, impedindo que o sangue extravase. Desde que os organismos estão sujeitos a sofrer traumatismos que podem romper vasos sangüíneos, o mecanismo de coagulação pode ser considerado como um fator de defesa natural.
Por outro lado, quando ocorrem perturbações no mecanismo da coagulação, mesmo lesões pequenas corno um corte superficial num dedo ou uma simples extração dentária podem provocar sangramentos intensos que duram horas ou dias, chegando a comprometer seriamente a vida do indivíduo.
Esses distúrbios no mecanismo de coagulação poderão ocorrer de forma inversa, ou seja, provocando coagulação anormal no interior dos vasos sangüíneos (trombose), fechando-os. Conseqüentemente, os tecidos servidos pelos vasos sangüíneos fechados sofrem falta de irrigação sangüínea e acabam por apresentar necrose tissular (morte do tecido).
Outra possibilidade é a de o coágulo, ou parte dele, destacar-se do local de sua formação, indo obstruir vasos sangüíneos situados em regiões mais distantes do organismo (fenômeno que caracteriza a chamada embolia), provocando nesses locais distúrbios circulatórios que, freqüentemente, levam o doente à morte.
Normalmente, o sangue em circulação é líquido, coagulando-se somente quando transborda dos vasos sangüíneos.
A fluidez do sangue no organismo depende dás propriedades físicas especiais do endotélio vascular (camada celular que reveste o interior dos vasos), da velocidade do fluxo sangüíneo, do número de células sangüíneas e da presença de anticoagulantes naturais, como a heparina, por exemplo.
Quando retirado do interior dos vasos, o sangue perde rapidamente sua fluidez, tornando-se inicialmente viscoso e adquirindo gradativamente consistência gelatinosa. Se uma pequena quantidade de sangue extravasar, em pouco tempo haverá formação de um coágulo semi-sólido.
De maneira simplificada, admite-se que o mecanismo de coagulação do sangue consiste em uma extensa reação em cadeia, na qual interferem diversas substâncias sangüíneas e celulares que agem umas sobre as outras, levando à formação de uma proteína especial, a fibrina, responsável final pelo processo de coagulação
4.1. FATORES ENVOLVIDOS: FATORES QUE PARTICIPAM DA COAGULAÇÃO DO SANGUE:
Fator I – fibrinogênio
Fator II - protrombina.
Fator III - trombopiastina.
Fator IV - íons-cálcio.
Fator V - pró-acelerina ou fator lábil.
Fator VI acelerina (participação duvidosa).
Fator VIl proconvertina ou fator estável
Fator VIII - globulina anti- hemofílica A.
Fator IX - globulina anti-hemofíiica B ou fator Christmas.
Fator X - fator Stuart-Prower.
Fator XI - precursor plasmático da tromboplastina.
Fator XII - fator Hageman.
Fator XIII fator estabilizador da fibrina
Apesar de o mecanismo da coagulação não ser completamente conhecido, existe uma teoria bastante difundida que atribui à coagulação a ação de doze fatores, indicados em algarismos romanos por convenção internacional.
A fase final da coagulação é determinada pela formação de fibrina, que se deposita sob a forma de um emaranhado de fios microscópicos, os quais acabam por aprisionar completamente as células sangüíneas. Os fios recém-formados aderem uns aos outros, às células do sangue, aos tecidos e à superfície alterada do revestimento interno dos vasos.- está formado o coágulo. O sangue extravasado transforma-se numa massa gelatinosa, interrompendo a hemorragia.
No entanto, esse é o final do processo de coagulação; para a formação de fibrina é necessário que todos os outros fatores tenham exercido sua atividade.
A fibrina resulta da transformação do fibrinogênio, proteína diluída no plasma (parte líquida do sangue) sangüíneo. Mas, para que o fibrinogênio se transforme, é necessária a intervenção da tromba que, por sua vez, é o resultado da transformação da protrombina, uma proteína (globulina) formada no ligado.
A responsável pela transformação da protrombina é a tromboplastina, substância presente nos tecidos e no interior das plaquetas (pequenos fragmentos celulares que se originam de grandes células da medula vermelha dos ossos, os megacariócitos). Quando a tromboplastina é liberada, inicia-se o processo de coagulação.
4.2. ALGUMAS ALTERAÇÕES DA COAGULAÇÃO
A falta de fibrinogênio poderá ser uma complicação séria da gravidez. Neste último caso, a deficiência de fibrinogênio pode ser resultado da entrada na circulação materna de líquido amniótico, rico em troplastina, ou de alguma alteração da placenta.
Pessoas que apresentam diminuição da quantidade de proteínas no plasma (hipoproteinemia), causada por um processo de desnutrição, ou, nos casos de doenças sangüíneas, uma menor quantidade de plaquetas presentes no sangue também poderão mostrar deficiências do processo de coagulação.
Outra doença temível que afeta a coagulação é a hemofilia enfermidade hereditária que, transmitida apenas pelas mulheres, atinge somente os homens. O indivíduo portador sofre durante toda a vida uma exagerada tendência a hemorragias, pois seu sangue coagula-se muito lentamente. Essa anormalidade apresenta-se sob três formas, conforme o fator coagulante deficiente:
Hemofilia A
Hemofilia B
Hemofilia C
5. O QUE É HEMOFILIA?
A Hemofilia é um distúrbio hereditário de coagulação sanguínea, na maioria dos casos ligada ao cromossomo X, que se manifestam quase exclusivamente nos indivíduos do sexo masculino. Aproximadamente 1 em cada 8.000 homens sofre da doença, que se caracteriza por uma anormalidade das proteínas funcionais de coagulação do plasma conhecidas como fatores VIII, IX e XI. Na maior parte dos casos, apisoas hemofílica apresenta quantidades normais do fator circulante deficiente, mas este está em estado funcionalmente inadequado. As pessoas com hemofilia têm um sangramento mais prolongado do que aquelas em níveis normais dos fatores VIII, IX, e XI em funcionamento, porém o funcionamento não é mais rápido do que ocorreria em uma pessoa com a mesma lesão. O sangramento pode ser externo, se a pele é danificada por um corte ou abrasão, ou pode ser interno, em músculos, articulações ou órgãos. A hemofilia é classificada de acordo com o fator de coagulação deficiente: se o fator deficiente é o VIII, temos a hemofilia A, se o fator é o IX, a hemofilia B e se o XI a hemofilia apresentada é a C. A hemofilia A é a mais comum, ocorrendo em 90% dos casos.
5.1. HISTÓRIA
Existe registro da hemofilia no sagrado texto judeu, o Talmud (cerca de 50-130dC). Segundo o Talmud uma criança não deveria ser circuncidada se já tivessem morrido dois irmãos em tal procedimento. No século XII, o médico árabe Abulcasis descreveu um caso de uma família cujos homens haviam morrido de sangramento após pequenos ferimentos. Em 1803, o médico norte-americano John Conrad constatou que havia "tendências a sangramentos em algumas famílias". Ele chegou à conclusão que a doença era hereditária e acometia mais homens do que mulheres.
O termo hemofilia apareceu pela primeira vez em 1828 por Hopff da Universidade de Zurique. Em 1937, Patek e Taylor, dois médicos de Harvard descobriram a globulina anti-hemofílica. Pavlosky, um médico de Buenos Aires, separou a Hemofilia A e Hemofilia B laboratorialmente.
Este teste era feito transferindo o sangue de um hemofílico para outro hemofílico. O fato corrigia o sangramento, comprovando que havia mais de um tipo de hemofilia. A Hemofilia é, muitas vezes, associada à história da Monarquia na Europa. A Rainha Vitória do Reino Unido passou a doença ao seu filho Leopoldo, e através de várias das suas filhas, a várias famílias reais Europeias, incluindo as famílias reais da Espanha, Alemanha, e Rússia. Alexei Romanov, filho to Czar Nicolau II da Rússia, foi um dos descendentes da Rainha Vitória que herdou a doença.
Durante as décadas de 1970 e 1980 a falta de outras modalidades de tratamento e a insuficiência de tecnologia para diagnosticar alguns vírus como o da AIDS acarretou em contaminação em grande escala da população hemofílica que dependiam de constantes transfusões de sangue. Mas este é um cenário que não está totalmente no passado, a maioria absoluta, em torno de 75%, da população mundial de hemofílicos não dispõe dos medicamentos básicos para o tratamento desta patologia, ficando ainda, estes, sujeitos às transfusões de sangue que pode lhes salvar a vida como também pode tirá-la.
5.2. GENÉTICA
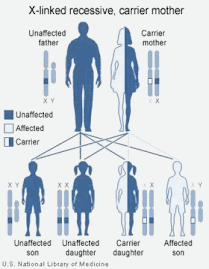
Um casal com um homem sadio e uma mulher portadora podem ter uma criança do sexo masculino sadia e uma hemofílico, ou uma criança do sexo feminino sadia e outra portadora.
A hemofilia, exceto sua variante "C", é referida como uma doença recessiva ligada ao cromossomo X ("doença ligada ao sexo"), o que significa que o gene defeituoso está localizado no cromossomo feminino ou cromossomo X.
Um homem possui um cromossomo X e um Y. Uma mulher, dois X. Como o defeito está no cromossomo X, é raro uma mulher que carregue o defeito, pois seu outro cromossomo X pode produzir os fatores de coagulação necessários. Entretanto, o cromossomo Y do homem não tem genes para os fatores de coagulação, portanto, se um homem apresentar defeito no cromossomo X, ele desenvolverá a doença.
Desde que um homem recebe o seu cromossomo X da mãe, o filho de uma portadora silênciosa tem 50% de chance de ter a doença e 50% de chance de ser sadio. Uma mulher para desenvolver a doença precisa receber dois cromossomos X defeituosos, um do pai e outro da mãe. Por isso a doença é mais comum em homens do que em mulheres. Entretanto há a possibilidade da mulher portadora silênciosa desenvolver uma hemofilia leve devido a lionização (inativação de um cromossomo X)
5.3. TIPOS DE HEMOFILIA
5.3.1 HEMOFILIA A (DEFICIÊNCIA DO FATOR VIII)
É a mais comum dos distúrbios hereditários com sangramento grave. É causada pela redução na quantidade ou na atividade do fator VIII. Essa proteína atua como co-fator para a ativação do fator X na cascata da coagulação. A hemofilia A é herdada como caráter recessivo ligado ao cromossomo X, conseqüentemente, ocorre em indivíduos do sexo masculino e indivíduos homozigotos do sexo feminino. A hemofilia A apresenta uma extensa gravidade clínica, que se correlaciona adequadamente com o nível de atividade do fato VIII. Dosagem de fator VIII pode classificar a hemofilia A em:
Grave: se o nível está abaixo de 1% do normal.
Moderada: se o nível está entre 2 a 5% do normal.
Leve: se o nível está entre 6 a 50% do normal.
5.3.2 HEMOFILIA B (DEFICIÊNCIA DO FATOR IX)
É uma doença causada pela deficiência grave do fato IX, apresenta-se de forma semelhante à hemofilia A, com exceção da inversão do DNA. Além disso a hemofilia B é também herdada como caráter recessivo ligada ao cromossomo X, podendo ocorrer de modo assintomático ou com hemorragia associada. Aproximadamente em 14% desses pacientes, o fator IX está presente, porém não é funcional.
É Também conhecida como doença Christmas, nome deriva do primeiro paciente descrito com essa afecção (e não do natal). A identificação dessa patologia só é possível através de ensaios dos níveis do fator.
5.3.3 HEMOFILIA C (DEFICIÊNCIA DO FATOR XI)
É também conhecida como Síndrome de Rosenthal, á causada pela deficiência do fator XI. Esse tipo de hemofilia costuma ser de nível leve, mas pode assemelhar-se à hemofilia clássica. Acomete ambos os sexos. É achado freqüente entre famílias judias descendentes de linhagens de judeus Asquenazi.
Difere da hemofilia A e B pois não leva a sangramentos nas juntas. Costuma cursar com sangramentos tardios em pós-operatório. É uma doença de herança autossômica dominante.
5.4 SINAIS E SINTOMAS CLÍNICOS
5.4.1 HEMARTROSE AGUDA
Aura, formigamento ou sensação de pontadas
Rigidez na posição de conforto
Limitação de amplitude de movimento
Dor
Edema
Sensibilidade
Calor
Hemartrose é o sangramento dentro dos espaços articulares, é uma das manifestações clínicas mais comuns da hemofilia. Pode resultar de um trauma ou estresse identificável ou pode ser espontâneo, mais freqüentemente afetando o joelho, cotovelo, tornozelo, quadril e ombro (ordem mais comum de aparecimento).
A hemartrose recorrente resulta em artropatia hemofílica (doença articular), com perda progressiva de movimento, atrofia muscular e contraturas em flexão. Os episódios de sangramento devem ser tratados precocemente com reposição de fator e imobilização da articulação durante o período de dor.
As hemartroses não são comuns no primeiro ano de vida, mas aumentam de freqüência logo que a criança começa a andar. A severidade da hemartrose pode variar (dependendo do grau da lesão), de dor suave e inchaço, os quais, se resolvem sem tratamento dentro de 1 a 3 dias, a dor severa com articulação inchada e dor martirizaste que persiste por muitos dias e apresenta resposta vagarosa ao tratamento.
5.4.2 HEMORRAGIA MUSCULAR
Dor se intensificando gradualmente
Espasmo protetor do músculo
Limitação do movimento nas articulações adjacentes
Músculo adota a posição de conforto (usualmente encurtado)
Perda de sensibilidade
O sangramento nos músculos é o segundo local de sangramento mais comum nas pessoas com hemofilia. As hemorragias musculares, podem ser mais insidiosa e massivas, que as hemorragias articulares. Elas podem ocorrer em qualquer local, mas são comuns nos grupos de músculos flexores, predominantemente o iliopsoas, o grastrocnêmio, e a superfície flexora do antebraço, e resulta em deformidades, tais como contraturas em flexão do quadril, posição eqüina do pé ou deformidade de Volkmann do antebraço.Quando o sangramento no músculo psoas ou ilíaco exerce pressão no ramo do nervo femoral que supre a pele na face anterior da coxa, ocorre uma perda de sensibilidade.
5.4.3 ENVOLVIMENTO GASTRINTESTINAL
Dor abdominal
Melena (sangue nas fezes)
Hematêmese (vomitar sangue)
Febre
Dor no abdômen inferior/virilha, devido ao sangramento da parede do intestino grosso ou do músculo iliopsoas.
Contratura de flexão do quadril, devido ao espasmo do músculo iliopsoas secundário a hemorragia.
A distensão dos músculos com sangue causa dor que pode ser sentida no abdômen inferior, possivelmente imitando até uma apendicite quando o sangramento ocorre no lado direito. Em uma tentativa de aliviar a distensão e reduzir a dor, pode ser adotada uma posição em flexão dos quadris.
Com o passar do tempo, as seguintes complicações podem ocorrer: Compressão vascular causando isquemia e necrose localizada;Substituição de fibras musculares por tecido fibrótico não-elástico causando encurtamento dos músculos e assim produzindo contraturas articulares;Lesões nervosas periféricas com compressão de um nervo localizado no mesmo compartimento que o hematoma, mais freqüentemente afetando os nervos femoral, ulnar e mediano,Formação de pseudotumor com erosão óssea.
5.5 DIAGNÓSTICO LABORATORIAL
Um exame de Tempo de tromboplastina parcial ativada (ou TTPA) prolongado com tempo de protrombina (ou TP) e tempo de coagulação normal deve ser investigado. Realizados através da adição de substâncias especiais ao sangue em estudo, seguido do registro do tempo de coagulação. Assim, pode-se realizar o exame de tempo de coagulação, que consiste na colocação de uma pequena quantidade de sangue em tubos especiais para se verificar quanto tempo decorre até a coagulação total.
Outro exame, constituído por uma picada no lóbulo da orelha, permite avaliar o tempo de sangramento. Nos casos em que existe tendência a hemorragias, administram-se drogas, como a vitamina k, que favorece a síntese da protrombina.
5.6 USO TERAPÊUTICO
5.6.1 O QUE É CONCENTRADO DE FATOR DE COAGULAÇÃO?
O concentrado do fator de coagulação ou “fator” é o fator concentrado transformado em pó. É diluído com água destilada para ficar líquido de novo e aplicado na veia. Alguns pacientes apresentaram a produção de anticorpos contra este concentrado.
Alguns concentrados de fatores são derivados do plasma, feitos a partir do sangue humano de doadores de sangue. Outros são fatores recombinantes, feitos em laboratório e não que contém sangue humano.
Algumas pessoas com hemofilia leve têm uma outra opção de tratamento que é o DDAVP – Desmopressina – medicação sintética, que não é derivada do sangue. O médico que acompanha o caso deverá orientar se é indicado usar o DDAVP.
5.6.2 QUANDO TRATAR?
Você deve ser tratado o mais rapidamente possível após uma lesão; se receber o fator logo após ter começado um sangramento, irá parar mais rapidamente e menos sangue terá para ser reabsorvido. Desta forma você voltará a sua rotina normal mais rapidamente. Se há uma dúvida sobre tratar uma hemorragia ou não, sempre decida pelo lado do tratamento: na dúvida, use fator.
5.7 O CENTRO DE TRATAMENTO DE HEMOFILIA (CTH).
Os Centros de Tratamento de Hemofilia (CTH), Serviços ou Unidades de Hemofilia são centros especializados em proporcionar tratamento multidisciplinar (com profissionais de várias áreas) para pessoas com distúrbios hemorrágicos.
Há vários CTH no Brasil e a maioria deles está nos Hemocentros, alguns em hospitais universitários ou outros. Os CTH tem em um mesmo lugar diagnóstico e acompanhamento por profissionais capacitados e experientes. Os CTH mais completos têm médicos hematologistas, ortopedistas, fisiatras, enfermeiros dentistas, fisioterapeutas, psicólogos, assistentes sociais e outros, além de dispensarem os fatores de coagulação, que no Brasil são adquiridos pelo Ministério da Saúde. A Federação Mundial de Hemofilia aconselha que o atendimento à pessoa com hemofilia seja feito por uma equipe multidisciplinar, já que se sabe que com essa abordagem os pacientes vivem mais e melhor.
5.8 CUIDADOS
Cuidados de enfermagem: * Proteger de traumatismos; * Imobilizar as articulações em casos de hemorragias articulares; * Observar e anotar episódios hemorrágicos; * Adotar cuidados especiais na realização de tricotomias, lavagens intestinais, aplicação de calor; * Auxiliar na higiene oral, atentando para não machucar a gengiva e mucosa oral; * Providenciar cartão de identificação do hemofílico, que deverá conter: grupo sangüíneo, fator Rh, pessoa a ser avisada em caso de urgência, nome do médico e endereço do hospital em que faz tratamento.
5.9 HEMOFILIA NO BRASIL
No Brasil, o fator VIII é fornecido gratuitamente pelo ministério da saúde por intermédio dos Hemocentros ou CTAs e graças a isso, os hemofílicos brasileiros estão tendo uma boa assistência apesar de as doses dispensadas serem ainda, apenas para tratamento dos eventos hemorrágicos e profilaxia em cirurgias sendo que a melhor maneira de tratar o paciente hemofílico sejam as doses profiláticas, administradas três vezes por semana, capazes de prevenir eficazmente as lesões articulares e outras complicações. Já existem estudos para implantação da dose profilática. Alguns hemofílicos graves, podem ainda desenvolver inibidores, que são anticorpos contra o fator VIII, dificultando o controle dos eventos hemorrágicos, mas que, felizmente, são poucos que desenvolvem e o MS dispõe também do fator VIIa para tratamento desses pacientes.
Podemos citar como exemplo personalidades importantes no Brasil, como o cartunista Henfil e seu irmão, o cientista social Betinho, entre tantos outros anônimos, foram vítimas deste tipo de ocorrência. O dia 17 de Abril é considerado o Dia do Hemofílico.
Herbert José de Souza, conhecido como Betinho, tornou-se reconhecido como sociólogo e ativista dos direitos humanos, sobretudo pelo seu projeto Ação da Cidadania contra a Miséria e pela Vida. Mineiro de Bocaiúva, nasceu dia 3 de novembro de 1935. Assim como seus irmãos Henfil e Chico Mário herdou da mãe a hemofilia. Em 1986, ao descobrir que contraiu o vírus da Aids em uma das transfusões de sangue a que se submetia freqüentemente, criou movimentos em defesa dos direitos dos portadores do vírus. Seus irmãos, Henfil e Chico Mário também morreram em 1988, em conseqüência da doença.
Betinho tornou-se reconhecido pelo movimento que liderou Ação da Cidadania contra Miséria e Pela Vida, em favor dos pobres e excluídos. Casado com Irles Carvalho e, pela segunda vez, com Maria Nakano. Morreu em 1997 e deixou dois filhos.
Frase de Betinho
"O Brasil tem fome de ética e passa fome em conseqüência da falta de ética na política.
domingo, 30 de março de 2008
1 COAGULAÇÃO DO SANGUE
O organismo conta com um mecanismo vital contra as perdas excessivas de sangue - a coagulação -, que auxilia na interrupção das hemorragias fechando os vasos sangüíneos abertos e, portanto, impedindo que o sangue extravase. Desde que os organismos estão sujeitos a sofrer traumatismos que podem romper vasos sangüíneos, o mecanismo de coagulação pode ser considerado como um fator de defesa natural.
Por outro lado, quando ocorrem perturbações no mecanismo da coagulação, mesmo lesões pequenas corno um corte superficial num dedo ou uma simples extração dentária podem provocar sangramentos intensos que duram horas ou dias, chegando a comprometer seriamente a vida do indivíduo.
Esses distúrbios no mecanismo de coagulação poderão ocorrer de forma inversa, ou seja, provocando coagulação anormal no interior dos vasos sangüíneos (trombose), fechando-os. Conseqüentemente, os tecidos servidos pelos vasos sangüíneos fechados sofrem falta de irrigação sangüínea e acabam por apresentar necrose tissular (morte do tecido).
Outra possibilidade é a de o coágulo, ou parte dele, destacar-se do local de sua formação, indo obstruir vasos sangüíneos situados em regiões mais distantes do organismo (fenômeno que caracteriza a chamada embolia), provocando nesses locais distúrbios circulatórios que, freqüentemente, levam o doente à morte.
Normalmente, o sangue em circulação é líquido, coagulando-se somente quando transborda dos vasos sangüíneos.
A fluidez do sangue no organismo depende dás propriedades físicas especiais do endotélio vascular (camada celular que reveste o interior dos vasos), da velocidade do fluxo sangüíneo, do número de células sangüíneas e da presença de anticoagulantes naturais, como a heparina, por exemplo.
Quando retirado do interior dos vasos, o sangue perde rapidamente sua fluidez, tornando-se inicialmente viscoso e adquirindo gradativamente consistência gelatinosa. Se uma pequena quantidade de sangue extravasar, em pouco tempo haverá formação de um coágulo semi-sólido.
De maneira simplificada, admite-se que o mecanismo de coagulação do sangue consiste em uma extensa reação em cadeia, na qual interferem diversas substâncias sangüíneas e celulares que agem umas sobre as outras, levando à formação de uma proteína especial, a fibrina, responsável final pelo processo de coagulação
2 FATORES ENVOLVIDOS
FATORES QUE PARTICIPAM DA COCOAGULAÇÃO DO SANGUE COAGULAÇÃO
Fator I - fibrinogênio.
Fator II - protrombina.
Fator III - trombopiastina.
Fator IV - íons-cálcio.
Fator V - pró-acelerina ou fator lábil.
Fator VI acelerina (participação duvidosa).
Fator VIl proconvertina ou fator estável
Fator VIII - globulina anti- hemofílica A.
Fator IX - globulina anti-hemofíiica B ou fator Christmas.
Fator X - fator Stuart-Prower.
Fator XI - precursor plasmático da tromboplastina.
Fator XII - fator Hageman.
Fator XIII fator estabilizador da fibrina
Apesar de o mecanismo da coagulação não ser completamente conhecido, existe uma teoria bastante difundida que atribui à coagulação a ação de doze fatores, indicados em algarismos romanos por convenção internacional.
A fase final da coagulação é determinada pela formação de fibrina, que se deposita sob a forma de um emaranhado de fios microscópicos, os quais acabam por aprisionar completamente as células sangüíneas. Os fios recém-formados aderem uns aos outros, às células do sangue, aos tecidos e à superfície alterada do revestimento interno dos vasos.- está formado o coágulo. O sangue extravasado transforma-se numa massa gelatinosa, interrompendo a hemorragia. No entanto, esse é o final do processo de coagulação; para a formação de fibrina é necessário que todos os outros fatores tenham exercido sua atividade.
A fibrina resulta da transformação do fibrinogênio, proteína diluída no plasma (parte líquida do sangue) sangüíneo. Mas, para que o fibrinogênio se transforme, é necessária a intervenção da tromba que, por sua vez, é o resultado da transformação da protrombina, uma proteína (globulina) formada no ligado.
A responsável pela transformação da protrombina é a tromboplastina, substância presente nos tecidos e no interior das plaquetas (pequenos fragmentos celulares que se originam de grandes células da medula vermelha dos ossos, os megacariócitos). Quando a tromboplastina é liberada, inicia-se o processo de coagulação.
3 ALGUMAS ALTERAÇÕES DA COAGULAÇÃO
A falta de fibrinogênio poderá ser uma complicação séria da gravidez. Neste último caso, a deficiência de fibrinogênio pode ser resultado da entrada na circulação materna de líquido amniótico, rico em troplastina, ou de alguma alteração da placenta.
Pessoas que apresentam diminuição da quantidade de proteínas no plasma (hipoproteinemia), causada por um processo de desnutrição, ou, nos casos de doenças sangüíneas, uma menor quantidade de plaquetas presentes no sangue também poderão mostrar deficiências do processo de coagulação.
Outra doença temível que afeta a coagulação é a hemofilia enfermidade hereditária que, transmitida apenas pelas mulheres, atinge somente os homens. O indivíduo portador sofre durante toda a vida uma exagerada tendência a hemorragias, pois seu sangue coagula-se muito lentamente. Essa anormalidade apresenta-se sob três formas, conforme o fator coagulante deficiente:
Hemofilia A
Hemofilia B
Hemofilia C
4 O QUE É HEMOFILIA?
A Hemofilia é um distúrbio hereditário de coagulação sanguínea, na maioria dos casos ligada ao cromossomo X, que se manifestam quase exclusivamente nos indivíduos do sexo masculino. Aproximadamente 1 em cada 8.000 homens sofre da doença, que se caracteriza por uma anormalidade das proteínas funcionais de coagulação do plasma conhecidas como fatores VIII, IX e XI. Na maior parte dos casos, apisoas hemofílica apresenta quantidades normais do fator circulante deficiente, mas este está em estado funcionalmente inadequado. As pessoas com hemofilia têm um sangramento mais prolongado do que aquelas em níveis normais dos fatores VIII, IX, e XI em funcionamento, porém o funcionamento não é mais rápido do que ocorreria em uma pessoa com a mesma lesão. O sangramento pode ser externo, se a pele é danificada por um corte ou abrasão, ou pode ser interno, em músculos, articulações ou órgãos. A hemofilia é classificada de acordo com o fator de coagulação deficiente: se o fator deficiente é o VIII, temos a hemofilia A, se o fator é o IX, a hemofilia B e se o XI a hemofilia apresentada é a C. A hemofilia A é a mais comum, ocorrendo em 90% dos casos.
5 HISTÓRIA
Existe registro da hemofilia no sagrado texto judeu, o Talmud (cerca de 50-130dC). Segundo o Talmud uma criança não deveria ser circuncidada se já tivessem morrido dois irmãos em tal procedimento. No século XII, o médico árabe Abulcasis descreveu um caso de uma família cujos homens haviam morrido de sangramento após pequenos ferimentos. Em 1803, o médico norte-americano John Conrad constatou que havia "tendências a sangramentos em algumas famílias". Ele chegou à conclusão que a doença era hereditária e acometia mais homens do que mulheres.
O termo hemofilia apareceu pela primeira vez em 1828 por Hopff da Universidade de Zurique. Em 1937, Patek e Taylor, dois médicos de Harvard descobriram a globulina anti-hemofílica. Pavlosky, um médico de Buenos Aires, separou a Hemofilia A e Hemofilia B laboratorialmente. Este teste era feito transferindo o sangue de um hemofílico para outro hemofílico. O fato corrigia o sangramento, comprovando que havia mais de um tipo de hemofilia.
A Hemofilia é, muitas vezes, associada à história da Monarquia na Europa. A Rainha Vitória do Reino Unido passou a doença ao seu filho Leopoldo, e através de várias das suas filhas, a várias famílias reais Europeias, incluindo as famílias reais da Espanha, Alemanha, e Rússia. Alexei Romanov, filho to Czar Nicolau II da Rússia, foi um dos descendentes da Rainha Vitória que herdou a doença.
Durante as décadas de 1970 e 1980 a falta de outras modalidades de tratamento e a insuficiência de tecnologia para diagnosticar alguns vírus como o da AIDS acarretou em contaminação em grande escala da população hemofílica que dependiam de constantes transfusões de sangue. Mas este é um cenário que não está totalmente no passado, a maioria absoluta, em torno de 75%, da população mundial de hemofílicos não dispõe dos medicamentos básicos para o tratamento desta patologia, ficando ainda, estes, sujeitos às transfusões de sangue que pode lhes salvar a vida como também pode tirá-la.
6 GENÉTICA
Um casal com um homem sadio e uma mulher portadora podem ter uma criança do sexo masculino sadia e uma hemofílico, ou uma criança do sexo feminino sadia e outra portadora.
A hemofilia, exceto sua variante "C", é referida como uma doença recessiva ligada ao cromossomo X ("doença ligada ao sexo"), o que significa que o gene defeituoso está localizado no cromossomo feminino ou cromossomo X.
Um homem possui um cromossomo X e um Y. Uma mulher, dois X. Como o defeito está no cromossomo X, é raro uma mulher que carregue o defeito, pois seu outro cromossomo X pode produzir os fatores de coagulação necessários. Entretanto, o cromossomo Y do homem não tem genes para os fatores de coagulação, portanto, se um homem apresentar defeito no cromossomo X, ele desenvolverá a doença.
Desde que um homem recebe o seu cromossomo X da mãe, o filho de uma portadora silênciosa tem 50% de chance de ter a doença e 50% de chance de ser sadio. Uma mulher para desenvolver a doença precisa receber dois cromossomos X defeituosos, um do pai e outro da mãe. Por isso a doença é mais comum em homens do que em mulheres. Entretanto há a possibilidade da mulher portadora silênciosa desenvolver uma hemofilia leve devido a lionização (inativação de um cromossomo X)
7 TIPOS DE HEMOFILIA
7.1 HEMOFILIA A (DEFICIÊNCIA DO FATOR VIII)
É a mais comum dos distúrbios hereditários com sangramento grave. É causada pela redução na quantidade ou na atividade do fator VIII. Essa proteína atua como co-fator para a ativação do fator X na cascata da coagulação. A hemofilia A é herdada como caráter recessivo ligado ao cromossomo X, conseqüentemente, ocorre em indivíduos do sexo masculino e indivíduos homozigotos do sexo feminino. A hemofilia A apresenta uma extensa gravidade clínica, que se correlaciona adequadamente com o nível de atividade do fato VIII. Dosagem de fator VIII pode classificar a hemofilia A em:
Grave: se o nível está abaixo de 1% do normal.
Moderada: se o nível está entre 2 a 5% do normal.
Leve: se o nível está entre 6 a 50% do normal.
7.2 HEMOFILIA B (DEFICIÊNCIA DO FATOR IX)
É uma doença causada pela deficiência grave do fato IX, apresenta-se de forma semelhante à hemofilia A, com exceção da inversão do DNA. Além disso a hemofilia B é também herdada como caráter recessivo ligada ao cromossomo X, podendo ocorrer de modo assintomático ou com hemorragia associada. Aproximadamente em 14% desses pacientes, o fator IX está presente, porém não é funcional. É Também conhecida como doença Christmas, nome deriva do primeiro paciente descrito com essa afecção (e não do natal). A identificação dessa patologia só é possível através de ensaios dos níveis do fator.
7.3 HEMOFILIA C (DEFICIÊNCIA DO FATOR XI)
É também conhecida como Síndrome de Rosenthal, á causada pela deficiência do fator XI. Esse tipo de hemofilia costuma ser de nível leve, mas pode assemelhar-se à hemofilia clássica. Acomete ambos os sexos. É achado freqüente entre famílias judias descendentes de linhagens de judeus Asquenazi.
Difere da hemofilia A e B pois não leva a sangramentos nas juntas. Costuma cursar com sangramentos tardios em pós-operatório. É uma doença de herança autossômica dominante.
8 SINAIS E SINTOMAS CLÍNICOS
8.1 HEMARTROSE AGUDA
Aura, formigamento ou sensação de pontadas
Rigidez na posição de conforto
Limitação de amplitude de movimento
Dor
Edema
Sensibilidade
Calor
Hemartrose é o sangramento dentro dos espaços articulares, é uma das manifestações clínicas mais comuns da hemofilia. Pode resultar de um trauma ou estresse identificável ou pode ser espontâneo, mais freqüentemente afetando o joelho, cotovelo, tornozelo, quadril e ombro (ordem mais comum de aparecimento).
A hemartrose recorrente resulta em artropatia hemofílica (doença articular), com perda progressiva de movimento, atrofia muscular e contraturas em flexão. Os episódios de sangramento devem ser tratados precocemente com reposição de fator e imobilização da articulação durante o período de dor.
As hemartroses não são comuns no primeiro ano de vida, mas aumentam de freqüência logo que a criança começa a andar. A severidade da hemartrose pode variar (dependendo do grau da lesão), de dor suave e inchaço, os quais, se resolvem sem tratamento dentro de 1 a 3 dias, a dor severa com articulação inchada e dor martirizaste que persiste por muitos dias e apresenta resposta vagarosa ao tratamento.
8.2 HEMORRAGIA MUSCULAR
Dor se intensificando gradualmente
Espasmo protetor do músculo
Limitação do movimento nas articulações adjacentes
Músculo adota a posição de conforto (usualmente encurtado)
Perda de sensibilidade
O sangramento nos músculos é o segundo local de sangramento mais comum nas pessoas com hemofilia. As hemorragias musculares, podem ser mais insidiosa e massivas, que as hemorragias articulares. Elas podem ocorrer em qualquer local, mas são comuns nos grupos de músculos flexores, predominantemente o iliopsoas, o grastrocnêmio, e a superfície flexora do antebraço, e resulta em deformidades, tais como contraturas em flexão do quadril, posição eqüina do pé ou deformidade de Volkmann do antebraço.Quando o sangramento no músculo psoas ou ilíaco exerce pressão no ramo do nervo femoral que supre a pele na face anterior da coxa, ocorre uma perda de sensibilidade.
8.3 ENVOLVIMENTO GASTRINTESTINAL
Dor abdominal
Melena (sangue nas fezes)
Hematêmese (vomitar sangue)
Febre
Dor no abdômen inferior/virilha, devido ao sangramento da parede do intestino grosso ou do músculo iliopsoas.
Contratura de flexão do quadril, devido ao espasmo do músculo iliopsoas secundário a hemorragia
A distensão dos músculos com sangue causa dor que pode ser sentida no abdômen inferior, possivelmente imitando até uma apendicite quando o sangramento ocorre no lado direito. Em uma tentativa de aliviar a distensão e reduzir a dor, pode ser adotada uma posição em flexão dos quadris.
Com o passar do tempo, as seguintes complicações podem ocorrer:
Compressão vascular causando isquemia e necrose localizada
Substituição de fibras musculares por tecido fibrótico não-elástico causando encurtamento dos músculos e assim produzindo contraturas articulares
Lesões nervosas periféricas com compressão de um nervo localizado no mesmo compartimento que o hematoma, mais freqüentemente afetando os nervos femoral, ulnar e mediano
Formação de pseudotumor com erosão óssea
9 DIAGNÓSTICO LABORATORIAL
Um exame de Tempo de tromboplastina parcial ativada (ou TTPA) prolongado com tempo de protrombina (ou TP) e tempo de coagulação normal deve ser investigado. Realizados através da adição de substâncias especiais ao sangue em estudo, seguido do registro do tempo de coagulação. Assim, pode-se realizar o exame de tempo de coagulação, que consiste na colocação de uma pequena quantidade de sangue em tubos especiais para se verificar quanto tempo decorre até a coagulação total. Outro exame, constituído por uma picada no lóbulo da orelha, permite avaliar o tempo de sangramento. Nos casos em que existe tendência a hemorragias, administram-se drogas, como a vitamina k, que favorece a síntese da protrombina.
10 USO TERAPÊUTICO
10.1 O QUE É CONCENTRADO DE FATOR DE COAGULAÇÃO?
O concentrado do fator de coagulação ou “fator” é o fator concentrado transformado em pó. É diluído com água destilada para ficar líquido de novo e aplicado na veia. Alguns pacientes apresentaram a produção de anticorpos contra este concentrado.
Alguns concentrados de fatores são derivados do plasma, feitos a partir do sangue humano de doadores de sangue. Outros são fatores recombinantes, feitos em laboratório e não que contém sangue humano.
Algumas pessoas com hemofilia leve têm uma outra opção de tratamento que é o DDAVP – Desmopressina – medicação sintética, que não é derivada do sangue. O médico que acompanha o caso deverá orientar se é indicado usar o DDAVP.
10.2 QUANDO TRATAR?
Você deve ser tratado o mais rapidamente possível após uma lesão; se receber o fator logo após ter começado um sangramento, irá parar mais rapidamente e menos sangue terá para ser reabsorvido. Desta forma você voltará a sua rotina normal mais rapidamente. Se há uma dúvida sobre tratar uma hemorragia ou não, sempre decida pelo lado do tratamento: na dúvida, use fator.
11 O CENTRO DE TRATAMENTO DE HEMOFILIA (CTH).
Os Centros de Tratamento de Hemofilia (CTH), Serviços ou Unidades de Hemofilia são centros especializados em proporcionar tratamento multidisciplinar (com profissionais de várias áreas) para pessoas com distúrbios hemorrágicos. Há vários CTH no Brasil e a maioria deles está nos Hemocentros, alguns em hospitais universitários ou outros. Os CTH tem em um mesmo lugar diagnóstico e acompanhamento por profissionais capacitados e experientes. Os CTH mais completos têm médicos hematologistas, ortopedistas, fisiatras, enfermeiros dentistas, fisioterapeutas, psicólogos, assistentes sociais e outros, além de dispensarem os fatores de coagulação, que no Brasil são adquiridos pelo Ministério da Saúde. A Federação Mundial de Hemofilia aconselha que o atendimento à pessoa com hemofilia seja feito por uma equipe multidisciplinar, já que se sabe que com essa abordagem os pacientes vivem mais e melhor.
12 CUIDADOS
Cuidados de enfermagem: * Proteger de traumatismos; * Imobilizar as articulações em casos de hemorragias articulares; * Observar e anotar episódios hemorrágicos; * Adotar cuidados especiais na realização de tricotomias, lavagens intestinais, aplicação de calor; * Auxiliar na higiene oral, atentando para não machucar a gengiva e mucosa oral; * Providenciar cartão de identificação do hemofílico, que deverá conter: grupo sangüíneo, fator Rh, pessoa a ser avisada em caso de urgência, nome do médico e endereço do hospital em que faz tratamento.
13 HEMOFILIA NO BRASIL
No Brasil, o fator VIII é fornecido gratuitamente pelo ministério da saúde por intermédio dos Hemocentros ou CTAs e graças a isso, os hemofílicos brasileiros estão tendo uma boa assistência apesar de as doses dispensadas serem ainda, apenas para tratamento dos eventos hemorrágicos e profilaxia em cirurgias sendo que a melhor maneira de tratar o paciente hemofílico sejam as doses profiláticas, administradas três vezes por semana, capazes de prevenir eficazmente as lesões articulares e outras complicações. Já existem estudos para implantação da dose profilática. Alguns hemofílicos graves, podem ainda desenvolver inibidores, que são anticorpos contra o fator VIII, dificultando o controle dos eventos hemorrágicos, mas que, felizmente, são poucos que desenvolvem e o MS dispõe também do fator VIIa para tratamento desses pacientes.
Podemos citar como exemplo personalidades importantes no Brasil, como o cartunista Henfil e seu irmão, o cientista social Betinho, entre tantos outros anônimos, foram vítimas deste tipo de ocorrência. O dia 17 de Abril é considerado o Dia do Hemofílico.
Herbert José de Souza, conhecido como Betinho, tornou-se reconhecido como sociólogo e ativista dos direitos humanos, sobretudo pelo seu projeto Ação da Cidadania contra a Miséria e pela Vida. Mineiro de Bocaiúva, nasceu dia 3 de novembro de 1935. Assim como seus irmãos Henfil e Chico Mário, herdou da mãe a hemofilia.
Em 1986, ao descobrir que contraiu o vírus da Aids em uma das transfusões de sangue a que se submetia freqüentemente, criou movimentos em defesa dos direitos dos portadores do vírus. Seus irmãos, Henfil e Chico Mário também morreram em 1988, em conseqüência da doença.
Betinho tornou-se reconhecido pelo movimento que liderou Ação da Cidadania contra Miséria e Pela Vida, em favor dos pobres e excluídos. Casado com Irles Carvalho e, pela segunda vez, com Maria Nakano. Morreu em 1997 e deixou dois filhos.
Frase de Betinho
"O Brasil tem fome de ética e passa fome em conseqüência da falta de ética na política.
O organismo conta com um mecanismo vital contra as perdas excessivas de sangue - a coagulação -, que auxilia na interrupção das hemorragias fechando os vasos sangüíneos abertos e, portanto, impedindo que o sangue extravase. Desde que os organismos estão sujeitos a sofrer traumatismos que podem romper vasos sangüíneos, o mecanismo de coagulação pode ser considerado como um fator de defesa natural.
Por outro lado, quando ocorrem perturbações no mecanismo da coagulação, mesmo lesões pequenas corno um corte superficial num dedo ou uma simples extração dentária podem provocar sangramentos intensos que duram horas ou dias, chegando a comprometer seriamente a vida do indivíduo.
Esses distúrbios no mecanismo de coagulação poderão ocorrer de forma inversa, ou seja, provocando coagulação anormal no interior dos vasos sangüíneos (trombose), fechando-os. Conseqüentemente, os tecidos servidos pelos vasos sangüíneos fechados sofrem falta de irrigação sangüínea e acabam por apresentar necrose tissular (morte do tecido).
Outra possibilidade é a de o coágulo, ou parte dele, destacar-se do local de sua formação, indo obstruir vasos sangüíneos situados em regiões mais distantes do organismo (fenômeno que caracteriza a chamada embolia), provocando nesses locais distúrbios circulatórios que, freqüentemente, levam o doente à morte.
Normalmente, o sangue em circulação é líquido, coagulando-se somente quando transborda dos vasos sangüíneos.
A fluidez do sangue no organismo depende dás propriedades físicas especiais do endotélio vascular (camada celular que reveste o interior dos vasos), da velocidade do fluxo sangüíneo, do número de células sangüíneas e da presença de anticoagulantes naturais, como a heparina, por exemplo.
Quando retirado do interior dos vasos, o sangue perde rapidamente sua fluidez, tornando-se inicialmente viscoso e adquirindo gradativamente consistência gelatinosa. Se uma pequena quantidade de sangue extravasar, em pouco tempo haverá formação de um coágulo semi-sólido.
De maneira simplificada, admite-se que o mecanismo de coagulação do sangue consiste em uma extensa reação em cadeia, na qual interferem diversas substâncias sangüíneas e celulares que agem umas sobre as outras, levando à formação de uma proteína especial, a fibrina, responsável final pelo processo de coagulação
2 FATORES ENVOLVIDOS
FATORES QUE PARTICIPAM DA COCOAGULAÇÃO DO SANGUE COAGULAÇÃO
Fator I - fibrinogênio.
Fator II - protrombina.
Fator III - trombopiastina.
Fator IV - íons-cálcio.
Fator V - pró-acelerina ou fator lábil.
Fator VI acelerina (participação duvidosa).
Fator VIl proconvertina ou fator estável
Fator VIII - globulina anti- hemofílica A.
Fator IX - globulina anti-hemofíiica B ou fator Christmas.
Fator X - fator Stuart-Prower.
Fator XI - precursor plasmático da tromboplastina.
Fator XII - fator Hageman.
Fator XIII fator estabilizador da fibrina
Apesar de o mecanismo da coagulação não ser completamente conhecido, existe uma teoria bastante difundida que atribui à coagulação a ação de doze fatores, indicados em algarismos romanos por convenção internacional.
A fase final da coagulação é determinada pela formação de fibrina, que se deposita sob a forma de um emaranhado de fios microscópicos, os quais acabam por aprisionar completamente as células sangüíneas. Os fios recém-formados aderem uns aos outros, às células do sangue, aos tecidos e à superfície alterada do revestimento interno dos vasos.- está formado o coágulo. O sangue extravasado transforma-se numa massa gelatinosa, interrompendo a hemorragia. No entanto, esse é o final do processo de coagulação; para a formação de fibrina é necessário que todos os outros fatores tenham exercido sua atividade.
A fibrina resulta da transformação do fibrinogênio, proteína diluída no plasma (parte líquida do sangue) sangüíneo. Mas, para que o fibrinogênio se transforme, é necessária a intervenção da tromba que, por sua vez, é o resultado da transformação da protrombina, uma proteína (globulina) formada no ligado.
A responsável pela transformação da protrombina é a tromboplastina, substância presente nos tecidos e no interior das plaquetas (pequenos fragmentos celulares que se originam de grandes células da medula vermelha dos ossos, os megacariócitos). Quando a tromboplastina é liberada, inicia-se o processo de coagulação.
3 ALGUMAS ALTERAÇÕES DA COAGULAÇÃO
A falta de fibrinogênio poderá ser uma complicação séria da gravidez. Neste último caso, a deficiência de fibrinogênio pode ser resultado da entrada na circulação materna de líquido amniótico, rico em troplastina, ou de alguma alteração da placenta.
Pessoas que apresentam diminuição da quantidade de proteínas no plasma (hipoproteinemia), causada por um processo de desnutrição, ou, nos casos de doenças sangüíneas, uma menor quantidade de plaquetas presentes no sangue também poderão mostrar deficiências do processo de coagulação.
Outra doença temível que afeta a coagulação é a hemofilia enfermidade hereditária que, transmitida apenas pelas mulheres, atinge somente os homens. O indivíduo portador sofre durante toda a vida uma exagerada tendência a hemorragias, pois seu sangue coagula-se muito lentamente. Essa anormalidade apresenta-se sob três formas, conforme o fator coagulante deficiente:
Hemofilia A
Hemofilia B
Hemofilia C
4 O QUE É HEMOFILIA?
A Hemofilia é um distúrbio hereditário de coagulação sanguínea, na maioria dos casos ligada ao cromossomo X, que se manifestam quase exclusivamente nos indivíduos do sexo masculino. Aproximadamente 1 em cada 8.000 homens sofre da doença, que se caracteriza por uma anormalidade das proteínas funcionais de coagulação do plasma conhecidas como fatores VIII, IX e XI. Na maior parte dos casos, apisoas hemofílica apresenta quantidades normais do fator circulante deficiente, mas este está em estado funcionalmente inadequado. As pessoas com hemofilia têm um sangramento mais prolongado do que aquelas em níveis normais dos fatores VIII, IX, e XI em funcionamento, porém o funcionamento não é mais rápido do que ocorreria em uma pessoa com a mesma lesão. O sangramento pode ser externo, se a pele é danificada por um corte ou abrasão, ou pode ser interno, em músculos, articulações ou órgãos. A hemofilia é classificada de acordo com o fator de coagulação deficiente: se o fator deficiente é o VIII, temos a hemofilia A, se o fator é o IX, a hemofilia B e se o XI a hemofilia apresentada é a C. A hemofilia A é a mais comum, ocorrendo em 90% dos casos.
5 HISTÓRIA
Existe registro da hemofilia no sagrado texto judeu, o Talmud (cerca de 50-130dC). Segundo o Talmud uma criança não deveria ser circuncidada se já tivessem morrido dois irmãos em tal procedimento. No século XII, o médico árabe Abulcasis descreveu um caso de uma família cujos homens haviam morrido de sangramento após pequenos ferimentos. Em 1803, o médico norte-americano John Conrad constatou que havia "tendências a sangramentos em algumas famílias". Ele chegou à conclusão que a doença era hereditária e acometia mais homens do que mulheres.
O termo hemofilia apareceu pela primeira vez em 1828 por Hopff da Universidade de Zurique. Em 1937, Patek e Taylor, dois médicos de Harvard descobriram a globulina anti-hemofílica. Pavlosky, um médico de Buenos Aires, separou a Hemofilia A e Hemofilia B laboratorialmente. Este teste era feito transferindo o sangue de um hemofílico para outro hemofílico. O fato corrigia o sangramento, comprovando que havia mais de um tipo de hemofilia.
A Hemofilia é, muitas vezes, associada à história da Monarquia na Europa. A Rainha Vitória do Reino Unido passou a doença ao seu filho Leopoldo, e através de várias das suas filhas, a várias famílias reais Europeias, incluindo as famílias reais da Espanha, Alemanha, e Rússia. Alexei Romanov, filho to Czar Nicolau II da Rússia, foi um dos descendentes da Rainha Vitória que herdou a doença.
Durante as décadas de 1970 e 1980 a falta de outras modalidades de tratamento e a insuficiência de tecnologia para diagnosticar alguns vírus como o da AIDS acarretou em contaminação em grande escala da população hemofílica que dependiam de constantes transfusões de sangue. Mas este é um cenário que não está totalmente no passado, a maioria absoluta, em torno de 75%, da população mundial de hemofílicos não dispõe dos medicamentos básicos para o tratamento desta patologia, ficando ainda, estes, sujeitos às transfusões de sangue que pode lhes salvar a vida como também pode tirá-la.
6 GENÉTICA
Um casal com um homem sadio e uma mulher portadora podem ter uma criança do sexo masculino sadia e uma hemofílico, ou uma criança do sexo feminino sadia e outra portadora.
A hemofilia, exceto sua variante "C", é referida como uma doença recessiva ligada ao cromossomo X ("doença ligada ao sexo"), o que significa que o gene defeituoso está localizado no cromossomo feminino ou cromossomo X.
Um homem possui um cromossomo X e um Y. Uma mulher, dois X. Como o defeito está no cromossomo X, é raro uma mulher que carregue o defeito, pois seu outro cromossomo X pode produzir os fatores de coagulação necessários. Entretanto, o cromossomo Y do homem não tem genes para os fatores de coagulação, portanto, se um homem apresentar defeito no cromossomo X, ele desenvolverá a doença.
Desde que um homem recebe o seu cromossomo X da mãe, o filho de uma portadora silênciosa tem 50% de chance de ter a doença e 50% de chance de ser sadio. Uma mulher para desenvolver a doença precisa receber dois cromossomos X defeituosos, um do pai e outro da mãe. Por isso a doença é mais comum em homens do que em mulheres. Entretanto há a possibilidade da mulher portadora silênciosa desenvolver uma hemofilia leve devido a lionização (inativação de um cromossomo X)
7 TIPOS DE HEMOFILIA
7.1 HEMOFILIA A (DEFICIÊNCIA DO FATOR VIII)
É a mais comum dos distúrbios hereditários com sangramento grave. É causada pela redução na quantidade ou na atividade do fator VIII. Essa proteína atua como co-fator para a ativação do fator X na cascata da coagulação. A hemofilia A é herdada como caráter recessivo ligado ao cromossomo X, conseqüentemente, ocorre em indivíduos do sexo masculino e indivíduos homozigotos do sexo feminino. A hemofilia A apresenta uma extensa gravidade clínica, que se correlaciona adequadamente com o nível de atividade do fato VIII. Dosagem de fator VIII pode classificar a hemofilia A em:
Grave: se o nível está abaixo de 1% do normal.
Moderada: se o nível está entre 2 a 5% do normal.
Leve: se o nível está entre 6 a 50% do normal.
7.2 HEMOFILIA B (DEFICIÊNCIA DO FATOR IX)
É uma doença causada pela deficiência grave do fato IX, apresenta-se de forma semelhante à hemofilia A, com exceção da inversão do DNA. Além disso a hemofilia B é também herdada como caráter recessivo ligada ao cromossomo X, podendo ocorrer de modo assintomático ou com hemorragia associada. Aproximadamente em 14% desses pacientes, o fator IX está presente, porém não é funcional. É Também conhecida como doença Christmas, nome deriva do primeiro paciente descrito com essa afecção (e não do natal). A identificação dessa patologia só é possível através de ensaios dos níveis do fator.
7.3 HEMOFILIA C (DEFICIÊNCIA DO FATOR XI)
É também conhecida como Síndrome de Rosenthal, á causada pela deficiência do fator XI. Esse tipo de hemofilia costuma ser de nível leve, mas pode assemelhar-se à hemofilia clássica. Acomete ambos os sexos. É achado freqüente entre famílias judias descendentes de linhagens de judeus Asquenazi.
Difere da hemofilia A e B pois não leva a sangramentos nas juntas. Costuma cursar com sangramentos tardios em pós-operatório. É uma doença de herança autossômica dominante.
8 SINAIS E SINTOMAS CLÍNICOS
8.1 HEMARTROSE AGUDA
Aura, formigamento ou sensação de pontadas
Rigidez na posição de conforto
Limitação de amplitude de movimento
Dor
Edema
Sensibilidade
Calor
Hemartrose é o sangramento dentro dos espaços articulares, é uma das manifestações clínicas mais comuns da hemofilia. Pode resultar de um trauma ou estresse identificável ou pode ser espontâneo, mais freqüentemente afetando o joelho, cotovelo, tornozelo, quadril e ombro (ordem mais comum de aparecimento).
A hemartrose recorrente resulta em artropatia hemofílica (doença articular), com perda progressiva de movimento, atrofia muscular e contraturas em flexão. Os episódios de sangramento devem ser tratados precocemente com reposição de fator e imobilização da articulação durante o período de dor.
As hemartroses não são comuns no primeiro ano de vida, mas aumentam de freqüência logo que a criança começa a andar. A severidade da hemartrose pode variar (dependendo do grau da lesão), de dor suave e inchaço, os quais, se resolvem sem tratamento dentro de 1 a 3 dias, a dor severa com articulação inchada e dor martirizaste que persiste por muitos dias e apresenta resposta vagarosa ao tratamento.
8.2 HEMORRAGIA MUSCULAR
Dor se intensificando gradualmente
Espasmo protetor do músculo
Limitação do movimento nas articulações adjacentes
Músculo adota a posição de conforto (usualmente encurtado)
Perda de sensibilidade
O sangramento nos músculos é o segundo local de sangramento mais comum nas pessoas com hemofilia. As hemorragias musculares, podem ser mais insidiosa e massivas, que as hemorragias articulares. Elas podem ocorrer em qualquer local, mas são comuns nos grupos de músculos flexores, predominantemente o iliopsoas, o grastrocnêmio, e a superfície flexora do antebraço, e resulta em deformidades, tais como contraturas em flexão do quadril, posição eqüina do pé ou deformidade de Volkmann do antebraço.Quando o sangramento no músculo psoas ou ilíaco exerce pressão no ramo do nervo femoral que supre a pele na face anterior da coxa, ocorre uma perda de sensibilidade.
8.3 ENVOLVIMENTO GASTRINTESTINAL
Dor abdominal
Melena (sangue nas fezes)
Hematêmese (vomitar sangue)
Febre
Dor no abdômen inferior/virilha, devido ao sangramento da parede do intestino grosso ou do músculo iliopsoas.
Contratura de flexão do quadril, devido ao espasmo do músculo iliopsoas secundário a hemorragia
A distensão dos músculos com sangue causa dor que pode ser sentida no abdômen inferior, possivelmente imitando até uma apendicite quando o sangramento ocorre no lado direito. Em uma tentativa de aliviar a distensão e reduzir a dor, pode ser adotada uma posição em flexão dos quadris.
Com o passar do tempo, as seguintes complicações podem ocorrer:
Compressão vascular causando isquemia e necrose localizada
Substituição de fibras musculares por tecido fibrótico não-elástico causando encurtamento dos músculos e assim produzindo contraturas articulares
Lesões nervosas periféricas com compressão de um nervo localizado no mesmo compartimento que o hematoma, mais freqüentemente afetando os nervos femoral, ulnar e mediano
Formação de pseudotumor com erosão óssea
9 DIAGNÓSTICO LABORATORIAL
Um exame de Tempo de tromboplastina parcial ativada (ou TTPA) prolongado com tempo de protrombina (ou TP) e tempo de coagulação normal deve ser investigado. Realizados através da adição de substâncias especiais ao sangue em estudo, seguido do registro do tempo de coagulação. Assim, pode-se realizar o exame de tempo de coagulação, que consiste na colocação de uma pequena quantidade de sangue em tubos especiais para se verificar quanto tempo decorre até a coagulação total. Outro exame, constituído por uma picada no lóbulo da orelha, permite avaliar o tempo de sangramento. Nos casos em que existe tendência a hemorragias, administram-se drogas, como a vitamina k, que favorece a síntese da protrombina.
10 USO TERAPÊUTICO
10.1 O QUE É CONCENTRADO DE FATOR DE COAGULAÇÃO?
O concentrado do fator de coagulação ou “fator” é o fator concentrado transformado em pó. É diluído com água destilada para ficar líquido de novo e aplicado na veia. Alguns pacientes apresentaram a produção de anticorpos contra este concentrado.
Alguns concentrados de fatores são derivados do plasma, feitos a partir do sangue humano de doadores de sangue. Outros são fatores recombinantes, feitos em laboratório e não que contém sangue humano.
Algumas pessoas com hemofilia leve têm uma outra opção de tratamento que é o DDAVP – Desmopressina – medicação sintética, que não é derivada do sangue. O médico que acompanha o caso deverá orientar se é indicado usar o DDAVP.
10.2 QUANDO TRATAR?
Você deve ser tratado o mais rapidamente possível após uma lesão; se receber o fator logo após ter começado um sangramento, irá parar mais rapidamente e menos sangue terá para ser reabsorvido. Desta forma você voltará a sua rotina normal mais rapidamente. Se há uma dúvida sobre tratar uma hemorragia ou não, sempre decida pelo lado do tratamento: na dúvida, use fator.
11 O CENTRO DE TRATAMENTO DE HEMOFILIA (CTH).
Os Centros de Tratamento de Hemofilia (CTH), Serviços ou Unidades de Hemofilia são centros especializados em proporcionar tratamento multidisciplinar (com profissionais de várias áreas) para pessoas com distúrbios hemorrágicos. Há vários CTH no Brasil e a maioria deles está nos Hemocentros, alguns em hospitais universitários ou outros. Os CTH tem em um mesmo lugar diagnóstico e acompanhamento por profissionais capacitados e experientes. Os CTH mais completos têm médicos hematologistas, ortopedistas, fisiatras, enfermeiros dentistas, fisioterapeutas, psicólogos, assistentes sociais e outros, além de dispensarem os fatores de coagulação, que no Brasil são adquiridos pelo Ministério da Saúde. A Federação Mundial de Hemofilia aconselha que o atendimento à pessoa com hemofilia seja feito por uma equipe multidisciplinar, já que se sabe que com essa abordagem os pacientes vivem mais e melhor.
12 CUIDADOS
Cuidados de enfermagem: * Proteger de traumatismos; * Imobilizar as articulações em casos de hemorragias articulares; * Observar e anotar episódios hemorrágicos; * Adotar cuidados especiais na realização de tricotomias, lavagens intestinais, aplicação de calor; * Auxiliar na higiene oral, atentando para não machucar a gengiva e mucosa oral; * Providenciar cartão de identificação do hemofílico, que deverá conter: grupo sangüíneo, fator Rh, pessoa a ser avisada em caso de urgência, nome do médico e endereço do hospital em que faz tratamento.
13 HEMOFILIA NO BRASIL
No Brasil, o fator VIII é fornecido gratuitamente pelo ministério da saúde por intermédio dos Hemocentros ou CTAs e graças a isso, os hemofílicos brasileiros estão tendo uma boa assistência apesar de as doses dispensadas serem ainda, apenas para tratamento dos eventos hemorrágicos e profilaxia em cirurgias sendo que a melhor maneira de tratar o paciente hemofílico sejam as doses profiláticas, administradas três vezes por semana, capazes de prevenir eficazmente as lesões articulares e outras complicações. Já existem estudos para implantação da dose profilática. Alguns hemofílicos graves, podem ainda desenvolver inibidores, que são anticorpos contra o fator VIII, dificultando o controle dos eventos hemorrágicos, mas que, felizmente, são poucos que desenvolvem e o MS dispõe também do fator VIIa para tratamento desses pacientes.
Podemos citar como exemplo personalidades importantes no Brasil, como o cartunista Henfil e seu irmão, o cientista social Betinho, entre tantos outros anônimos, foram vítimas deste tipo de ocorrência. O dia 17 de Abril é considerado o Dia do Hemofílico.
Herbert José de Souza, conhecido como Betinho, tornou-se reconhecido como sociólogo e ativista dos direitos humanos, sobretudo pelo seu projeto Ação da Cidadania contra a Miséria e pela Vida. Mineiro de Bocaiúva, nasceu dia 3 de novembro de 1935. Assim como seus irmãos Henfil e Chico Mário, herdou da mãe a hemofilia.
Em 1986, ao descobrir que contraiu o vírus da Aids em uma das transfusões de sangue a que se submetia freqüentemente, criou movimentos em defesa dos direitos dos portadores do vírus. Seus irmãos, Henfil e Chico Mário também morreram em 1988, em conseqüência da doença.
Betinho tornou-se reconhecido pelo movimento que liderou Ação da Cidadania contra Miséria e Pela Vida, em favor dos pobres e excluídos. Casado com Irles Carvalho e, pela segunda vez, com Maria Nakano. Morreu em 1997 e deixou dois filhos.
Frase de Betinho
"O Brasil tem fome de ética e passa fome em conseqüência da falta de ética na política.
segunda-feira, 20 de agosto de 2007
CoNsTrUçãO
Em Construção!!!
Por favor, tenham paciência que logo estaremos postando novos comentarios e reportagens!!!!
abração
Por favor, tenham paciência que logo estaremos postando novos comentarios e reportagens!!!!
abração
quinta-feira, 16 de agosto de 2007
quarta-feira, 1 de agosto de 2007
segunda-feira, 30 de julho de 2007
Tempo
"Tudo tem o seu tempo certo e há tempo para todo o propósito debaixo dos céus"(Ecl3.1)
Nunca esquecer:Deus traz a existência aquilo que não existe.
Assinar:
Postagens (Atom)